



")














- Adv -
La sindrome di Rothmund-Thomson (SRT), detta anche poichilodermia di Rothmund-Thomson o congenita, è una malattia rara di origine genetica che si manifesta nella prima infanzia e colpisce diverse parti del corpo.
I disturbi (sintomi) caratteristici della malattia furono descritti per la prima volta nel 1868 dall’oculista tedesco Auguste Rothmund e successivamente, nel 1921, dal dermatologo inglese Sydney Thomson. Nel 1957 il medico statunitense William Taylor ha accomunato i due casi denominando la malattia “sindrome di Rothmund-Thomson”.
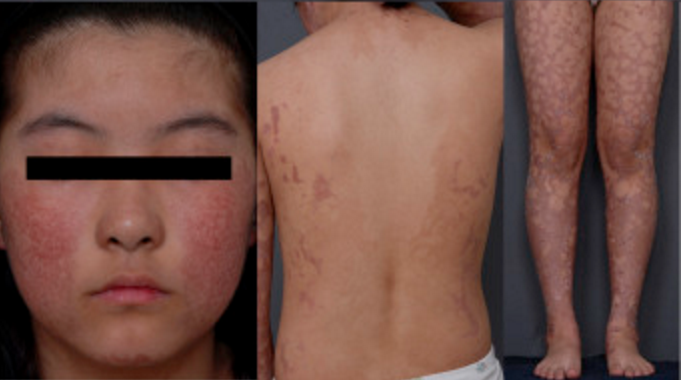
La SRT è caratterizzata da alterazioni della pelle quali atrofia, eccesso o carenza di pigmentazione e dilatazione dei capillari (complessivamente definite poichilodermia), associate a ritardo della crescita sia prima che dopo la nascita, bassa statura, capelli, ciglia e sopracciglia radi o assenti, cataratta giovanile, difetti scheletrici e dentali, invecchiamento precoce e aumentato rischio di sviluppare tumori, particolarmente alle ossa (sarcomi) e, in età più avanzata, alla pelle.
La frequenza della malattia nella popolazione generale non è nota ma, dalla prima ad oggi, i casi descritti nella letteratura medica sono meno di 500.
SINTOMATOLOGIA
La sindrome di Rothmund-Thomson (SRT) si manifesta nella prima infanzia. A 3-6 mesi di età comincia a svilupparsi un arrossamento sulle guance, simile a una scottatura da sole che, successivamente, si diffonde in altre parti del corpo, soprattutto gambe, braccia e glutei, mentre il tronco viene risparmiato. Col progredire della malattia si sviluppa l’atrofia della pelle con aree a forma di reticolo caratterizzate da eccesso e carenza di pigmentazione, vesciche e dilatazione dei capillari (teleangectasie) che diventano croniche (permanenti); tale quadro viene complessivamente definito poichilodermia.
Altre manifestazioni includono alterazioni delle unghie e ispessimento della pelle del palmo delle mani e dei piedi. Nel 50% circa dei bambini con SRT compare, mediamente tra i 2 e i 7 anni di età, la cataratta (opacizzazione del cristallino) giovanile bilaterale, ossia a entrambi gli occhi. Essa determina una grave compromissione della vista che può, tuttavia, essere recuperata con un immediato intervento chirurgico. Una larga percentuale di persone con SRT presenta, inoltre, un ritardo nella crescita e bassa statura associata a malformazioni delle ossa (fronte prominente, naso a sella, malformazione degli arti e delle mani) e dei denti e osteopenia, cioè ridotta massa ossea. In alcune persone sono stati descritti anche disturbi (sintomi) gastrointestinali e respiratori, alterazioni del sangue più o meno gravi (come l’alterata produzione delle cellule del sangue da parte del midollo) e riduzione della funzionalità degli organi sessuali (ipogonadismo).
Inoltre, le persone con SRT hanno un aumentato rischio di sviluppare, in particolare nell’infanzia, tumori, alle ossa e in epoca successiva alla pelle (carcinoma cutaneo a cellule squamose). L’età di comparsa, così come il tipo e la gravità dei sintomi, possono variare da caso a caso.
Complessivamente i segni caratteristici della malattia includono:
-
poichilodermia
-
ritardo della crescita
-
bassa statura
-
capelli, ciglia e sopracciglia radi o assenti
-
cataratta giovanile bilaterale
-
difetti scheletrici e dentali
-
invecchiamento precoce
-
aumentato rischio di sviluppare tumori
In base alla causa e alla varietà dei sintomi presenti, in aggiunta alla poichilodermia, sono state descritte due forme di SRT:
-
tipo 1 (SRT1), la cui causa genetica non è nota e che si manifesta principalmente con poichilodermia e cataratta giovanile bilaterale
-
tipo 2 (SRT2), causata nella maggioranza dei casi da mutazioni nel gene RECQL4; questa forma si manifesta con poichilodermia, malformazioni ossee e ritardo della crescita e presenta un aumentato rischio di sviluppare tumori



